Human Lymphoid Tissue Deconvolution with SpaRank
This tutorial shows how a single SpaRank model trained on a human lymphoid single-cell reference can be applied across multiple spatial sections spanning different tissue types (tonsil, lymph node, and spleen) and spatial transcriptomics platforms (Visium, CytAssist Visium, and Array-seq). Datasets are available on Zenodo.
[ ]:
import muon as mu
import os, glob
from os.path import join
import scanpy as sc
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import sparank
from sparank.config import ExpConfig, ModalityConfig, SimulationConfig
from sparank.framework import SpaRank
1. Data
We start by loading the single-cell reference used for training, which contains cells from three lymphoid tissues: tonsil, lymph node, and spleen.
[2]:
data_dir = '../data/human_lymphoid'
# single-cell reference data
adx_sc_rna = sc.read_h5ad(join(data_dir, 'sc/adx.h5ad'))
adx_sc_rna.var_names_make_unique()
2. Training
The cell below shows the training configuration used in the experiments reported in our paper. It is commented out by default, so you only need to run it if you want to reproduce the model from scratch.
A few settings are specific to this dataset:
context_key="Tissue": tissue identity is used as context, allowing one model to adapt across tonsil, lymph node, and spleen.cl_weight=0.5, mrp_weight=0.5: contrastive learning and reconstruction objectives are enabled to improve robustness.batch_key="Sample": marker genes are computed at the sample level for finer-grained feature selection.simulation.batch_key="Tissue": when simulating pseudo-spots for training, cells are sampled from the same tissue to preserve tissue-specific composition.
[3]:
# cfg = ExpConfig(
# modalities=[
# ModalityConfig(name="rna", top_k=500, cl_dropout_rate=0.3, mrp_mask_rate=0.3)
# ],
# simulation=SimulationConfig(
# total_samples=100_000,
# batch_request_size=100_000,
# cells_mean=10,
# cells_std=5,
# cell_sample_method='gaussian',
# batch_key='Tissue',
# ),
# celltype_key='Subset',
# batch_key='Sample',
# context_key='Tissue',
# dim=64,
# depth=1,
# heads=1,
# cl_weight=0.5,
# cl_temperature=0.1,
# mrp_weight=0.5,
# cls_weight=1.0,
# epochs=20,
# num_workers=4,
# batch_size=256
# )
# model = SpaRank(cfg=cfg, save_dir=f'../outputs/human_lymphoid')
# model.register_modality("rna", adx_sc_rna)
# model.prepare()
# model.fit()
# model.save()
3. Load pre-trained checkpoint
[4]:
model = SpaRank.load(f'../outputs/human_lymphoid', device="cuda:0")
4. Predict on spatial datasets
We apply the pretrained model to each spatial dataset in turn. Before prediction, we set the Tissue field in obs so the model receives the same tissue context used during training.
[5]:
import matplotlib.pyplot as plt
def show_pred(adx, preds, yinv=False, s=50):
ct_names = preds.columns.to_list()
adx.obs[[f'deconv_{x}' for x in ct_names]] = preds
# ad_plot = adx[adx.obs['ID_patients']==sample_key].copy()
ad_plot = adx.copy()
n_col = 3
n_row = len(ct_names) // n_col + 1
fig, axes = plt.subplots(n_row, n_col, figsize=(n_col * 4, n_row * 4))
for i, ct in enumerate(ct_names):
# 兼容 axes 扁平化处理
ax = axes.flat[i] if n_row > 1 else axes[i]
# 使用筛选后的 ad_plot 进行绘图
sc.pl.embedding(
ad_plot, # <--- 改用筛选后的对象
basis='spatial',
color=f'deconv_{ct}',
ax=ax,
show=False,
title=f'{ct}',
frameon=False,
s=s,
vmin=0, vmax='p99.2', cmap='magma'
)
if yinv:
ax.invert_yaxis()
# 隐藏多余的空图
for j in range(i + 1, len(axes.flat)):
axes.flat[j].axis('off')
plt.tight_layout()
# plt.savefig(f'{fig_save_dir}/deconv_{ep}.png')
plt.show()
4.1 Tonsil
We begin with a tonsil section and provide Tissue="Tonsil" before running inference.
[6]:
adx_tnsl = sc.read_h5ad('../data/human_lymphoid/sp/tonsil/adx_rna_rep1.h5ad')
adx_tnsl.obs['Tissue'] = 'Tonsil' # set context
tnsl_preds = model.predict(mod_adatas={'rna':adx_tnsl})
/opt/conda/envs/scvi-env/lib/python3.13/site-packages/sparank/data/dataset.py:198: UserWarning: To copy construct from a tensor, it is recommended to use sourceTensor.detach().clone() or sourceTensor.detach().clone().requires_grad_(True), rather than torch.tensor(sourceTensor).
torch.tensor(self.context_ids[idx], dtype=torch.long)
/opt/conda/envs/scvi-env/lib/python3.13/site-packages/sparank/data/dataset.py:198: UserWarning: To copy construct from a tensor, it is recommended to use sourceTensor.detach().clone() or sourceTensor.detach().clone().requires_grad_(True), rather than torch.tensor(sourceTensor).
torch.tensor(self.context_ids[idx], dtype=torch.long)
[7]:
show_pred(adx_tnsl, tnsl_preds)
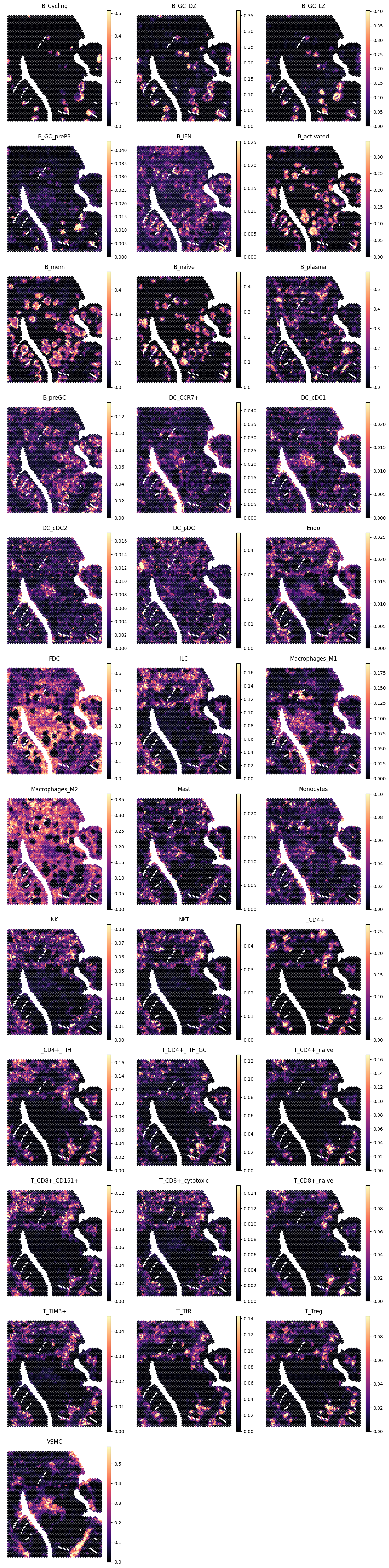
4.2 Lymph node (CytAssist Visium)
We next apply the same model to a lymph node section profiled with CytAssist Visium, using Tissue="LN" as context.
[8]:
adx_lnc = sc.read_h5ad('../data/human_lymphoid/sp/lymph_cyt_visium/adx_rna.h5ad')
adx_lnc.obs['Tissue'] = 'LN' # set context
lnc_preds = model.predict(mod_adatas={'rna':adx_lnc})
/opt/conda/envs/scvi-env/lib/python3.13/site-packages/sparank/data/dataset.py:198: UserWarning: To copy construct from a tensor, it is recommended to use sourceTensor.detach().clone() or sourceTensor.detach().clone().requires_grad_(True), rather than torch.tensor(sourceTensor).
torch.tensor(self.context_ids[idx], dtype=torch.long)
[9]:
show_pred(adx_lnc, lnc_preds)
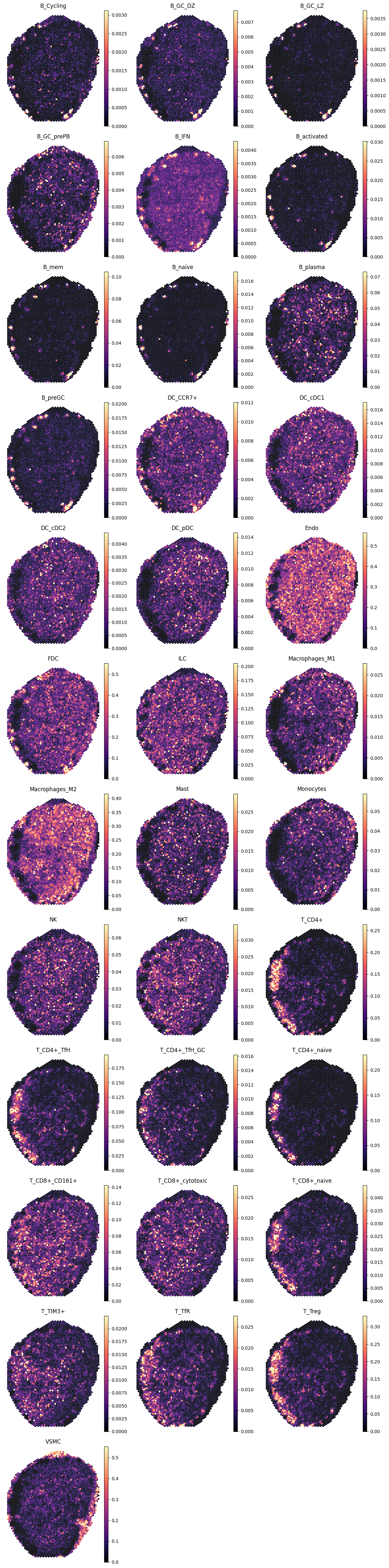
4.3 Lymph node (Visium)
[10]:
adx_lnv = sc.read_h5ad('../data/human_lymphoid/sp/lymph_visium/adx_rna.h5ad')
adx_lnv.obs['Tissue'] = 'LN' # set context
lnv_preds = model.predict(mod_adatas={'rna':adx_lnv})
/opt/conda/envs/scvi-env/lib/python3.13/site-packages/sparank/data/dataset.py:198: UserWarning: To copy construct from a tensor, it is recommended to use sourceTensor.detach().clone() or sourceTensor.detach().clone().requires_grad_(True), rather than torch.tensor(sourceTensor).
torch.tensor(self.context_ids[idx], dtype=torch.long)
[11]:
show_pred(adx_lnv, lnv_preds, yinv=True)
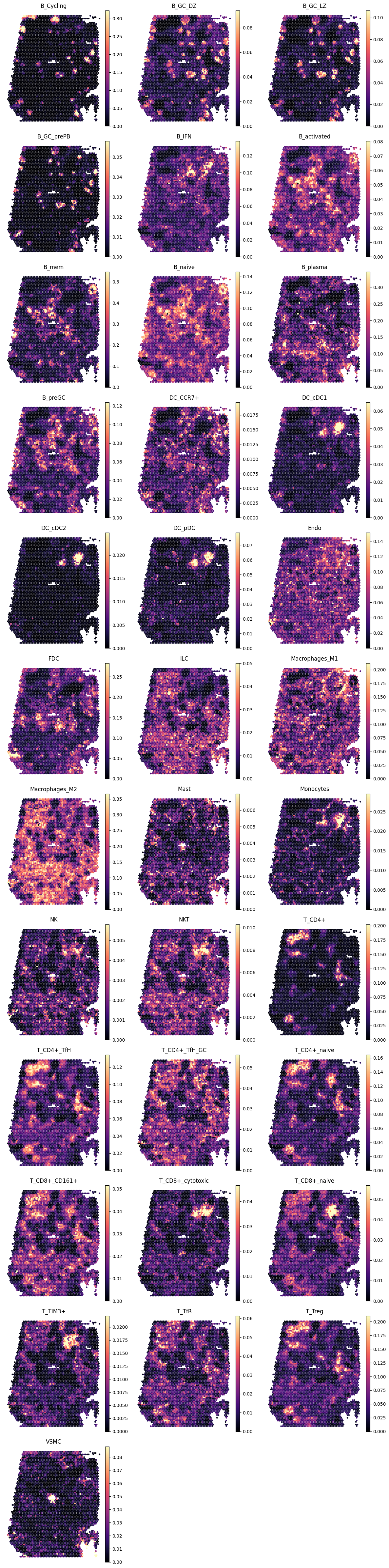
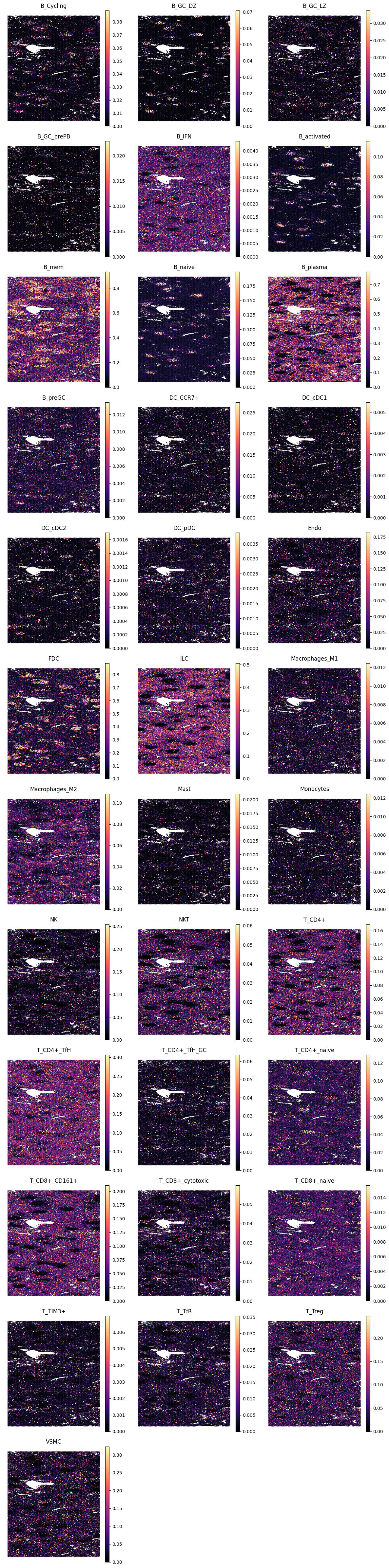
4.4 Spleen
For spleen, we apply preprocessing before inference. Broadly distributed genes, including highly abundant diffuse signals and selected immunoglobulin or mitochondrial genes, are removed first. We then keep highly variable genes to focus prediction on more informative spatial structure.
[12]:
def get_hvg(adx, n_top_genes=5000):
adata = adx.copy()
sc.pp.highly_variable_genes(adata, flavor='seurat_v3', n_top_genes=n_top_genes)
return adata.var.query('highly_variable').index.to_list()
[13]:
adx_spl = sc.read_h5ad('../data/human_lymphoid/sp/spleen/adx_rna.h5ad')
adx_spl.obs['Tissue'] = 'Spleen' # set context
## remove those genes that spread out the whole section
K = 100
high_gs = adx_spl.var['SYMBOL'][np.argpartition(adx_spl.X.sum(axis=0).A1, kth=-K)[-K:]]
adx_spl = adx_spl[:, ~adx_spl.var['SYMBOL'].isin(high_gs)].copy()
gs_mask = (~adx_spl.var['SYMBOL'].str.startswith('IG')) & (~adx_spl.var['SYMBOL'].str.startswith('MT-'))
adx_spl = adx_spl[:, gs_mask].copy()
hvgs = get_hvg(adx_spl, 8000)
adx_spl = adx_spl[:, hvgs].copy()
spl_preds = model.predict(mod_adatas={'rna':adx_spl})
/tmp/ipykernel_212061/368084282.py:6: FutureWarning: Series.__getitem__ treating keys as positions is deprecated. In a future version, integer keys will always be treated as labels (consistent with DataFrame behavior). To access a value by position, use `ser.iloc[pos]`
high_gs = adx_spl.var['SYMBOL'][np.argpartition(adx_spl.X.sum(axis=0).A1, kth=-K)[-K:]]
/opt/conda/envs/scvi-env/lib/python3.13/site-packages/sparank/data/dataset.py:198: UserWarning: To copy construct from a tensor, it is recommended to use sourceTensor.detach().clone() or sourceTensor.detach().clone().requires_grad_(True), rather than torch.tensor(sourceTensor).
torch.tensor(self.context_ids[idx], dtype=torch.long)
[14]:
show_pred(adx_spl, spl_preds, s=8)
[ ]: